


執筆:弁護士 早崎 智久(メディカル・ビューティー・ヘルスケアチーム)
連載:薬機法とは ~薬機法の基本~
『第1回 薬機法の全体像』はこちらから
『第2回 「医薬品等」とは』はこちらから
『第3回 医薬品の販売と薬局 ① -医薬品の種類-』はこちらから
『第4回 医薬品の販売と薬局② -医薬品の販売と薬局-』はこちらから
『第5回 医薬品の製造販売① -全体像と製造販売業・製造業・製造管理-』はこちらから
『第6回 医薬品の製造販売② -医薬品の製造販売承認-』はこちらから
『第7回 製造販売後の医薬品 -医薬品の安全管理と有効性の再確認-』はこちらから
『第8回 医薬品情報の消費者への表示-医薬品の表示、添付文書、広告-』はこちらから
【目次】
1 はじめに
2 医薬品の安全管理・有効性の再確認の制度の概要
⑴ 医薬品の問題点
⑵ 安全管理・有効性の再確認のための薬機法の制度
3 安全管理のための制度
⑴ 副作用等の情報の収集
⑵ 副作用等の情報の検討(分析、評価)と対策の決定
⑶ 対策の指示・指導
⑷ 医療機関、薬局における情報活用・対策
⑸ 医薬品の安全管理のまとめ
4 製造販売業者における市場流通後の安全管理
⑴ 安全管理制度における製造販売業者の役割
⑵ GVP省令の内容
⑶ GVP省令に基づく体制
⑷ 製造販売業者の種類に応じた体制の違い
5 有効性の再確認のための制度
⑴ 有効性の再確認のための制度の概要
⑵ 新医薬品等の再審査制度(薬機法第14条の4)
⑶ 医薬品の再評価制度(薬機法第14条の6)
⑷ GPSP省令
6 まとめ
1.はじめに
GVA法律事務所では、メディカル、美容、ヘルスケア領域に関して専門チームを設け、各分野について多様なサポートをさせていただいております。
本稿は薬機法の基本に関する連載になりますが、5回目と6回目の2回を通じて、「製造販売」という医薬品を市場に流通させることに関する規制をご説明しました。
7回目となる今回は、製造販売(市場に流通した)後の医薬品の安全管理と有効性の再確認について解説いたします。
2.医薬品の安全管理・有効性の再確認の制度の概要
前々回と前回、医薬品を市場に流通させるためには、医薬品が有効性、安全性、品質のいずれにおいても問題がないことを確認する必要があること、そのチェックのための制度を確認しました。
しかし、実際に医薬品を市場に流通させた後になってから、その問題点が判明することもあります。そのため、医薬品に関しては「市場に流通させたら終わり」というものではなく、市場に流通させた後の安全対策なども必要になります。
今回は、市場流通後の医薬品に関する諸制度を見てまいります。
⑴ 医薬品の問題点
市場流通後の諸制度を理解するためには、医薬品特有の問題点を確認することで非常に視点がクリアになります。そこで、まずは、医薬品特有の問題点を、①安全性、②品質、③有効性の3つの観点から確認しましょう。
① 安全性(副作用)
医薬品が、通常の「モノ」とは異なり、人体に対して使用するという特殊なものであることはこれまでにも繰り返しお話してきました。
医薬品には、効き目がある(有効性)だけでなく、安全性や品質も求められ、前回の医薬品の薬事承認においても、適切に非臨床試験や臨床試験を実施し、その際にはデータの正確性に限らず、データの完全性、つまり、都合の悪いデータも含めて審査の対象になることも確認しました。特に、副作用の恐ろしさ、過去に起きてしまった薬害のような悲劇を防止するためにも、慎重な審査が求められています。
とはいえ、審査前の試験は、どれだけ慎重にやっても限界があります。市場に流通した後は、何万人という数の患者さんが使用することになる医薬品も、試験の時点では多くても数百人というレベルで治験を行うことが限界になります。
さらに、治験では、公正に検証するための客観的なデータを得るために、使用の際の条件を設けています。つまり、治験に参加する患者さんの属性(年齢、症状の程度など)を統一させ、使用量なども制限します。しかし、市場流通後に医薬品が使用される場面では、各患者さんの年齢も、その症状の程度も異なりますので、各医師は、それぞれの患者さんに合わせて、用量を変えなければいけません。そのため、治験の時とは全く違う条件で使用されることになるため、数百人の治験では発生しない副作用が、後になって判明することもあり得るのです。
以上から、医薬品の安全性を高めるためには、市場流通後のチェック=安全管理が必要不可欠になります。
② 品質
また、有効性や安全性だけでなく、製造における品質管理も重要になります。
しっかりとした製法に基づき、製造工程のマニュアルが定められていても、実際の現場でいい加減な製造が行われてしまえば、問題のある医薬品が生み出されます。これが大きく問題になったのが、HIVウイルスに感染した血液製剤です。この事件では、血液製剤の原材料となった人間の血液がHIVに感染しており、血液製剤を必要としていた血友病の患者の方がHIVに感染してしまうという悲劇を生みました。その原因は、原材料の品質管理に大きな問題があったことが分かっています。
そのため、市場流通後も、医薬品の品質管理は常に必要になります。
③ 有効性
医薬品の有効性については、治験や非臨床試験において、データに基づく証明が必要になることを前回確認しました。とはいえ、この証明は永遠のものではありません。
例えば、ある医薬品が対象とする疾患についても、その医薬品よりも遥かに効き目のある新しい医薬品が開発されることがあります。より効き目があり、より安全な製品が登場すれば、古い医薬品が不必要になることもあり得ます。また、安全性と同様に、治験は限られた人数で行うだけなので、より多くの人が使用した結果、治験のデータとは異なり、あまり有効性がないことが判明することもあり得ます。また、その結果、想定された副作用とのバランスを考えた時に、医薬品として評価できなくなる場合もあります。
そのため、医薬品は、市場流通後も、有効性を再評価する必要があります。
⑵ 安全管理・有効性の再確認のための薬機法の制度
薬機法では、このような問題に対処するため、各種の制度を設けています。
そして、これらの制度は、市場流通後の医薬品に関する調査を前提にします。このような調査は、「製造販売後調査」(Postmarketing Surveillance=略称「PMS」(ピーエムエス))と呼ばれています。
まずは、各制度の概要を見ましょう。
① 安全管理のための制度
まず、安全管理のための制度ですが、大きくは以下のようなフローになっています。
つまり、医薬品の副作用などの安全性に関する情報を国に集まるようにし、国はこの情報をもとにしっかりと対策を検討して実施します。さらに、必要な情報は速やかに医療機関や国民に共有されるという流れです。
ⅰ 副作用等の情報の収集
ⅱ 副作用等の情報の検討(分析、評価)と対策の決定
ⅲ 厚生労働省による対策の指示・指導
ⅳ 医療機関、薬局における情報活用・対策
② 品質管理のための制度
次に品質を維持するための制度です。
これは、前々回にご説明した「品質管理」が該当します。薬事承認前にチェックされるものですが、製造販売後は、その基準を満たして継続的に運用していくこと、製造していくことが求められます。
③ 有効性の再確認のための制度
最後に、有効性の再確認のための制度です。
この制度は、新薬の場合と販売されてから長い年月が経っている医薬品の場合の2種類に分かれています。
ⅰ 新医薬品等の再審査制度(薬機法第14条の4)
ⅱ 医薬品の再評価制度(薬機法第14条の6)
ポイント:医薬品の問題点とこれに対する各種制度
① 医薬品の安全性の問題 ⇒ 市場流通後の安全管理の制度
② 医薬品の品質の問題 ⇒ 製造段階における品質管理の制度
③ 医薬品の有効性の問題 ⇒ 市場流通後の再審査制度、再評価制度
②に関しては本連載の第5回でご説明していますので、以下では、市場流通後に関する①と③の各制度について、詳細にご説明いたします。
3.安全管理のための制度
⑴ 副作用等の情報の収集
医薬品の安全性に問題があった場合の対処は、最終的に国(厚生労働省)が措置を実施することで行います。
しかし、これを実施するためには、副作用や感染症などの安全性に関する情報を国に集まるようにする必要があります。具体的には、そのような情報を国に報告させることが不可欠となります。
この情報収集のための制度として、以下のような制度が定められています。
副作用、感染症に関する情報収集のための主な制度
◎製造販売業者から
・企業報告制度(薬機法第68条の10第1項)
・感染症定期報告制度(薬機法第68条の24)
◎医療機関から
・医薬品・医療機器等安全性情報報告制度(薬機法第68条の10第2項)
◎患者から
・患者副作用報告制度
◎国際機関から
・WHO国際医薬品モニタリング制度
◎医薬品の種類に応じたもの
・新薬の市販直後調査(薬機法79条第1項…条件付与→GVP第10条第1項)
・要指導医薬品の製造販売後調査
まず、これらの情報収集の制度について、順番に解説します。
① 製造販売業者を通じた情報収集
…「企業報告制度」
この連載の前回と前々回において、医薬品の製造販売業者が医薬品の市場流通に関して責任を負うことを説明しましたが、市場流通後の安全性の確認もこれに含まれます。具体的には、製造販売業者は、製造販売する医薬品について、副作用と感染症に関する情報を収集し、これを厚生労働大臣に報告する義務があります。これを企業報告制度といいます。
ⅰ 製造販売業者による情報の収集(薬機法第68条の2の5第1項及び第2項)
薬機法第68条の2の5第1項は、製造販売業者が、医薬品の適正な使用のために必要な情報を収集して検討すると共に、この情報を、薬局、医療機関、販売業者等、医療従事者その他の医薬関係者に提供するものとしています。
また、同条第2項は、これらの医薬関係者は、製造販売業者の情報収集に協力するよう努力義務を課しています。
そのため、製造販売業者は、自社の医薬品の製造販売のための営業をすると共に、当該製品の安全性に関する情報を常に収集しなければならないことになります。薬機法の条文でも、医薬関係者がこれに協力すると定められているとおり、これらの情報収集は、主にそのような医療の現場で行われています。製造販売業者には、医薬情報担当者(Medical Representative=略称「MR」(エムアール))という従業員がおりますが、彼らは、一般的なイメージとなっている営業を行うだけでなく、現場において安全性に関する情報の収集も行っています。
さらに、製造販売業者は、このような現場での意見に限らず、国内外の学会や研究会、学術論文、専門誌などで発表される知見からも、安全性に関する情報を収集する必要があります。
ⅱ 厚生労働大臣への報告(薬機法第68条の10第1項)
製造販売業者は、収集・検討した情報が、医薬品の有効性・安全性において重要な事項の場合は、厚生労働大臣(窓口はPMDA)に報告する義務があります。
この報告の対象となる重要な事項について、薬機法第68条の10第1項は、「当該品目の副作用その他の事由によるものと疑われる疾病、障害又は死亡の発生、当該品目の使用によるものと疑われる感染症の発生その他の医薬品、医薬部外品、化粧品、医療機器又は再生医療等製品の有効性及び安全性に関する事項で厚生労働省令で定めるもの」とあります。
つまり、医薬品が原因だと疑われる副作用、感染症の発生などの医薬品の有効性・安全性に関する事項であり、具体的には、厚生労働省令で定めるとされています。
そして、この内容を定めているのが厚生労働省令=薬機法施行規則の第228条の20です。特に深刻な状況の場合の「一」と、ある程度の深刻な場合の「二」、それ以外の「三」と分けられています。
このうちの「一」は、情報を知った時から15日以内、「二」も30日以内、と短期間での報告を義務付けており、早期に国が情報を把握できるようになっています。
② 医療機関を通じた情報収集
…「医薬品・医療機器等安全性情報報告制度」
上記①の企業報告制度は製造販売業者が情報を収集して報告する制度ですが、医療機関も、この情報収集に協力するものとされています。もっとも、医療機関には、製造販売業者に協力するだけでなく、自らも厚生労働大臣(窓口はPMDA)に報告する義務があります。これを医薬品・医療機器等安全性情報報告制度といいます。
発生してしまった副作用の症状や感染症については、現場の医師らが実際に触れた事象なので、その内容をもっとも把握しているのも医師らです。この制度で収集される情報は、直接に関知した者による情報なので、安全管理のためのとても重要な情報になっています。
③ 患者からの情報収集
…「患者副作用報告制度」
副作用の影響を直接に受けるのは服用した患者本人です。そのため、PMDAでは、患者本人やその家族からの副作用報告を受け付けています。ここで受け付けられた情報は整理・検討され、厚生労働省に報告されます。
平成31年から始まった新しい制度ですが、更なる情報収集のスピードの向上などが期待されます。
④ 国際的な情報収集
…「WHO国際医薬品モニタリング制度」
WHO=世界保健機関は、国際連合に設置されている専門機関ですが、このWHOが国家間において副作用などの情報を共有するために発足させた制度が「WHO国際医薬品モニタリング制度」です。
前述の①~③の各制度が日本国内での情報共有と対応のための制度であったのに対し、国際医薬品モニタリング制度は、これを地球規模にしたものであり、参加国は自国で確認・検討された副作用や感染症に関する情報を報告し、その情報が参加国で共有されることにより、世界規模での情報の共有が可能となっています。
⑤ 医薬品の種類に応じた情報収集
ア 新薬
…「新薬の市販直後調査」
…「再審査制度」
前述したように、医薬品の副作用は、治験の対象者数が少ないことなどから、薬事承認時には確認できないものもあります。この場合は、市場流通後に副作用が判明していきますが、その可能性が最も高いのは、言うまでもなく新薬です。
そのため、新薬については、薬事承認の際に、「市販直後調査の実施」が条件として設定されます。これを「市販直後調査」といいます。
具体的には、販売後の6か月間が調査期間となりますが、新薬については、後述の要指導医薬品と同様に、通常の企業報告制度のほか重点的な調査が必要とされています。
なお、製造販売業者は、市場直後調査実施計画書を作成し、医療機関に調査への協力を求めることとされていますので、医療機関においても、新薬の取扱いにおいて慎重な対応を採ることができるようになっています。
さらに、新薬に関しては再審査制度も重要になりますが、この制度は、安全性のほか、有効性の再審査の意味もあるので、後述します。
イ 要指導医薬品
…「要指導医薬品の製造販売後調査」
要指導医薬品については、この連載の3回目において解説しましたが、その中には、医療用医薬品から要指導医薬品になったばかりのスイッチ直後品目や、新しい有効成分を含んだダイレクト直後品目が含まれています。
要指導医薬品はあくまで市販薬のため、消費者が自分の意思で選択して購入するものであり、医療用医薬品のように医師が管理して処方するものではありませんが、このような品目に関しては、上記アの新薬と同様に、副作用等の安全性については通常の医薬品以上に慎重な管理が必要になります。
この点、まず、ダイレクト直後品目は6か月間の市販直後調査が行われます(新薬と同じ)。次に、スイッチ直後品目は、まず原則3年間の製造販売後調査が行われ、その後の使用成績評価により、要指導医薬品から一般用医薬品の第一類医薬品となり、さらに第二類・第三類への区分替えの検討という流れになります。
⑥ 感染症に関する情報収集
…「感染症定期報告制度」
医薬品の原材料として生物由来の成分を使用することがあります。この場合、原材料となった成分が感染症に汚染されていると、医薬品の服用を通じて感染症が発生することになります(血液製剤を通じて、大きな悲劇が過去に起きたことはご説明したとおりです)。
そのため、副作用と同様に、感染症に関しても情報収集を行い、被害の拡大を食い止めることが必要不可欠です。
薬機法は、このための制度として、製造販売業者に定期報告の義務を課しています。つまり、製造販売業者は、副作用に関する企業報告制度、感染症に関する感染症定期報告制度という2つの報告義務があります。
なお、製造販売業者は、PMDAに定期的に報告を行い、この報告を通じて収集された情報は、厚生労働省、薬事・食品衛生審議会により分析、検討され、必要がある場合には対策が行われます。
⑵ 副作用等の情報の検討(分析、評価)と対策の決定
以上のように情報を集めるための様々な制度を見てきましたが、これらを通じて、国のもとには製造販売業者、医療機関、国際機関から医薬品の副作用や感染症などの安全性に関する情報が収集されます。
前述のとおり、製造販売業者からの情報はPMDAに報告されますので、まず、PMDAにおいて、収集された情報を整理し、調査検討を行います。この調査結果が厚生労働省に通知されると(薬機法68条の13第4項)、厚生労働省は、これを薬事・食品衛生審議会に報告し、必要がある場合には意見を求めます(薬機法68条の12)。
薬事・食品衛生審議会の医薬品安全対策部会は、検討の上で、厚生労働大臣に対して意見を述べ、その内容を踏まえ、厚生労働大臣は対策を決定することになります。
この場合に、厚生労働省が検討するのは、以下のような事項です。
ⅰ 緊急性が高い場合
→ 医薬品がこれ以上流通しないように、販売停止・回収などが必要かどうか
→ 医療機関などに情報を緊急に伝える必要があるかどうか
→ 使用上の注意の改訂を命じ、医療機関などに周知させる必要があるかどうか
ⅱ 比較的緊急性が高くない場合
→ 厚生労働省ではなく、製造販売業者に自主的な措置をさせるかどうか
ⅲ 更なる調査が必要な場合
→ 更に情報収集をするかどうか
⑶ 対策の指示・指導
上記の検討を経て対策が決定されると、次はこれを実施することになりますが、取られる対策には以下のようなものがあります。
① 製造販売業者に対する行政措置・行政指導
医薬品の安全性に問題がある場合には、被害の拡大を防ぐため、危険を除去する必要があります。そのため、厚生労働省は、製造販売業者などに対して、製品の回収、出荷の停止、販売の停止などを命じることになります。また、違反の程度によっては、製造販売の承認の取り消しがなされることもあります。
② 医療機関、薬局、消費者への情報提供
安全性に問題があるとされた医薬品ですが、この医薬品自体はすでに市場に流通しています。また、医薬品ごとに安全性への問題の程度も異なりますので、その程度に応じて、医薬品を処方する医師や薬局、使用する患者などの消費者に向けて、情報を提供することが極めて重要になります。
このための制度として、以下のように様々なものがあります。
ア 緊急安全性情報(イエローレター)
緊急安全性情報、通称「イエローレター」は、特に緊急性が高い場合に提供されるものです。具体的には、副作用の影響により服用者は死亡に至るような場合や副作用が発生する可能性が非常に高い場合です。
情報の発出者は製造販売業者ですが、安全対策措置の一環としての情報提供になりますので、厚生労働省の関与のもとでPMDAと協議の上でイエローレターは作成されます。通称通り、背景が黄色の書面(データ)であり、周囲を太い赤で囲んだものなので、見た目でも緊急性が強いことが示されています。
極めて深刻な場合に出されるものなので、頻出されるものではありませんが、直近では、社会問題にもなったタミフル服用後の異常行動に関するものが2007年3月に出されています。
イ 安全性速報(ブルーレター)
イエローレターに較べると緊急性は高くないものの、注意喚起を行うべき事案で提供されるのが安全性速報、通称「ブルーレター」です。文字通り、背景が青色の書面(データ)であり、周囲を赤い太で囲んだものです。本日時点においては、過去17年間で15件が発出されています。
なお、情報の発出者が製造販売業者であり、厚生労働省の関与のもとでPMDAと協議の上で作成されることはイエローレターと同様です。

イエローレターの例 ブルーレターの例
※いずれも厚生労働省ホームページ「医薬品等安全性関連情報」の掲載情報から引用
ウ 「使用上の注意」の改訂
詳細は次回にご説明しますが、医薬品には添付文書が必要です。そこには、医薬品の服用方法や副作用などの重要な情報のほか、「使用上の注意」などが記載されていることは、皆さんにもお馴染みかと思います。
そして、安全性に関して問題があれば、その内容は「使用上の注意」などとして、追記する必要があるのが一般的です。そのため、厚生労働省の対策の一つとして、製造販売業者は医薬品の使用上の注意を改訂し、これを公表します。この内容は、厚生労働省のホームページでも随時公表されていますが、2021年度は計19件、2022年度は本日時点で計14件の改訂が行われており、医薬品の安全性対策が常に行われていることが実感されます。
エ 医薬品・医療機器等安全性情報
「医薬品・医療機器等安全性情報」は、厚生労働省の生活衛生局が、定期的に公表しているものであり、上記ア~ウやその他の安全性に関する情報をまとめて公表するものです。内容は上記と重複するものもありますが、各月の安全対策を詳細な内容を含めて一覧的に確認できるものとして、有意義な情報提供になっています。
オ PMDAによる各種情報提供
PMDAは、厚生労働省の指示のもとで安全対策のための情報の整理・検討を行っており、安全性に関する多くの情報を扱っています。
そのため、PMDAのホームページの最上段に安全性に関する情報ページへのリンクが貼られており、そちらのページで、上記ア~エはもちろんのこと、リスクのある案件を含めた多量の情報を公開しています。
医薬品に限らず、医薬品等(医薬品、医療機器、医薬部外品、化粧品、再生医療等製品)の全ての安全性に関する情報を確認できるものとなっています。
その他、あまり一般的に知られているとまではいえないですが、「患者向け医薬品ガイド」というものが秀逸です。これは、膨大な医薬品のそれぞれごとに、使用方法、使用上の注意などの医薬品に関する詳細な情報が記載されています。そのため、万が一添付文書を亡くしてしまったような場合もこちらから情報を確認できますし、添付文書の小さな文字が読みにくい方は、こちらを確認するほうが可読性にも優れています。
なお、こちらも次回にご説明しますが、令和3年の薬機法改正により、医療用医薬品の添付文書は電子化され、PMDAによって管理されています。
⑷ 医療機関、薬局における情報活用・対策
当然のことですが、厚生労働省やPMDAが情報を広く提供する目的は、この情報を受け取った医療機関や消費者が、その情報に従って、特定の医薬品の使用を止めたり、注意して使用するなどの対応を求めるためです。
そのため、薬機法第68条の2の5第3項では、医療機関などに対して「情報の活用」を求めています。つまり、国が公表した情報の内容を踏まえて、患者への処方を止める、処方を変える、使用方法を変えるなどし、また、患者に情報を伝えるなどの適切な対処をするように努力する義務があるものとしています。
⑸ 医薬品の安全管理のまとめ
以上、医薬品の安全対策の制度を見てきましたが、PMDAがこれを分かりやすく図にしたものが以下です。
安全性に関する情報が収集され、整理・検討を経て、対応策が決定・実施され、併せて医療機関や国民に情報提供されることで、医薬品の安全対策が実施されていることがお分かりになるかと思います。
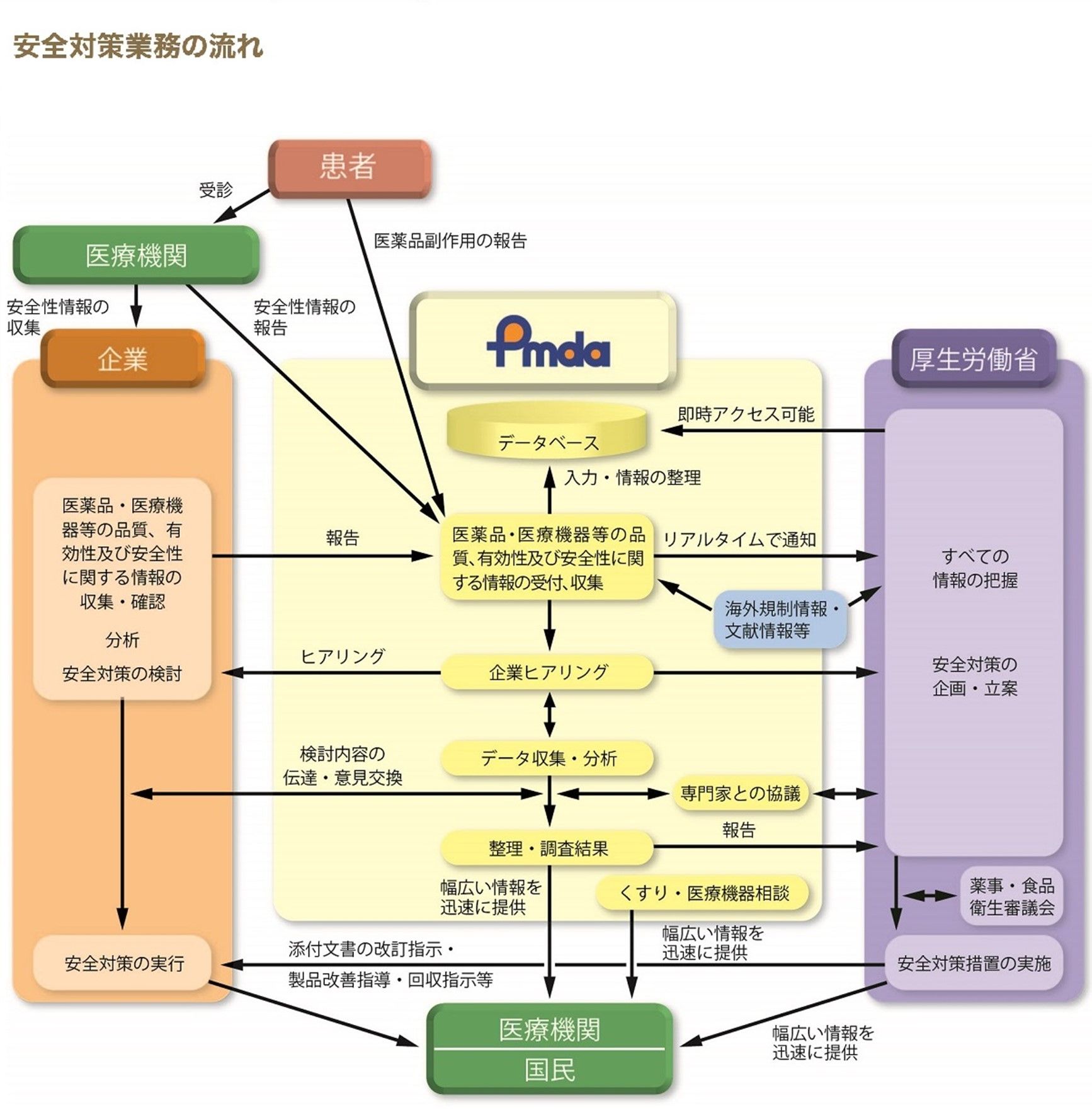
独立行政法人医薬品医療機器総合機構ホームページから引用
4.製造販売業者における市場流通後の安全管理
⑴ 安全対策制度における製造販売業者の役割
上記の「3」では、医薬品が市場に流通した後の、PMSを通じた安全管理の制度をご説明しました。そして、これらの制度では、製造販売業者の果たす役割が大きく、製造販売業者が医薬品に関する責任を負うということが前提になっています。
そして、本連載の製造販売業者に関する回で触れた事項の復習になりますが、
・製造販売業許可を得るためには、①医薬品の「品質管理」と、②医薬品の「市場流通後の安全管理」の2つが必要になる
・許可の基準も、申請者がこの2つのことをできるかどうかという点から行われる
ということをご説明しました。
そして、この許可の基準は、以下の厚生労働省令としてまとめられていることも確認しました。
① 品質管理の基準
=『医薬品、医薬部外品、化粧品及び再生医療等製品の品質管理の基準に関する省令』
(通称「GQP省令」=Good Quality Practice)
② 製造販売後の安全管理の基準
=『医薬品、医薬部外品、化粧品、医療機器及び再生医療等製品の製造販売後安全管理の基準に関する省令』
(通称「GVP省令」=Good Vigilance Practice)
この内の①については、その際にご説明しましたが、②は名称のとおり、製造販売後の安全管理に関するものとなりますので、今回、ご説明します。
(なお、再掲ですが、GVPの「V」は、「(安全性を)警戒・用心(Vigilance)して管理する」ということになります。)
安全管理のための諸制度は、医薬品が市場に流通、つまり、製造販売業者の手元を離れた場面での事項になりますが、GVP省令が定めるのは、その安全管理のための制度において製造販売業者が担う役割、義務を適切に果たさせるための事項になります。GVP省令に沿った体制がつくられ、この体制のもとに対応が取られることで、安全管理制度は初めて機能するものと言えます。
⑵ GVP省令の内容
GVP省令に関しては、製造販売業者の許可の基準を定める薬機法第12条の2の第2項において、安全対策の内容に関して「‥‥医薬品…の製造販売後安全管理(品質、有効性及び安全性に関する事項その他適正な使用のために必要な情報の収集、検討及びその結果に基づく必要な措置をいう。以下同じ。)の方法」と定めています。
つまり、製造販売業者は、市場流通後の医薬品について、品質・有効性・安全性に必要な情報の収集、検討及びその結果に基づく必要な措置を採ることができるようにするよう求められているということになります。ここでいう「必要な情報の収集、検討及びその結果に基づく必要な措置」というのは、前述の安全管理の制度で触れたことと同じことです。
つまり、GVP省令は、製造販売業者に対して、前述の「安全管理を実施できる組織体制であることを求めている基準」ということになります。
⑶ GVP省令に基づく体制
このように、安全対策は、GVP省令が定める基準に沿った組織で行われますが、この役割を中心になって担うのが、製造販売業者のなかの安全管理部門(後述する第一種製造販売業者に関しては、「安全管理統括部門」)であり、そのトップは「三役」の一人である「安全管理責任者」になります。
安全管理統括部門は、製造販売後安全管理手順書という安全管理のためのマニュアルを作成し、これに基づき、安全管理情報の収集や検討を行います。そして、問題があれば、安全を確保するために必要な措置を立案した上で、これを統括製造販売責任者に報告します。統括製造販売責任者がこの報告の内容を確認(評価)し、その指示のもとで、安全管理統括部門は措置を実施することになります。
この場合、当然のことですが、安全管理部門と製造部門(外部の製造業者の場合も多い)との間の連携が必要になります。この場合、製造販売業者内で製造業者を監督するのは品質保証部門になりますので、統括の監督のもと、両部門が緊密に連携を取ることが必要になります。
⑷ 製造販売業者の種類に応じた体制の違い
なお、医薬品には様々な種類があることは、本連載の第3回目にご説明しましたが、そのリスクに応じて、製造販売業者に求められる責任の大きさにも違いがあります。
この違いは、医薬品の種類に応じる形で表れており、処方箋医薬品は第一種医薬品製造販売業者、処方箋医薬品以外の医薬品(=その他の医療用医薬品と一般用医薬品)は第二種製造販売業者となります。
第一種製造販売業者の場合は、安全管理責任者の資格要件(3年以上の経験を有する者)があり、自ら業務を行う場合を除き、安全管理実施責任者の設置も求められます。
5.有効性の再確認のための制度
市場流通後の医薬品の安全性の問題に続き、次は、市場流通後の医薬品の有効性の問題に関する制度を見ていきましょう。
⑴ 有効性の再確認のための制度の概要
前述のように、医薬品の有効性が問題になる場面は大きく分けて2パターンあります。
1つ目は、新薬の場合です。新薬の場合は、治験を経ただけでまだ市場では使用されていませんので、治験で服用した患者の数はごく少数です。そのため、安全性の問題に加えて、有効性についてもケースが少なく、前述した治験の性質もあって、有効性に関しても、承認時点とは異なることがあります。このため、市場流通後にデータを収集し、これに基づいて改めて審査する必要があります。これを「再審査制度」といいます。
2つ目は、販売されてから長い期間を経た医薬品の場合です。長い期間を経たということは、その間に、他の医薬品が販売され、医学や薬学が発展しているということでもあります。つまり、その医薬品に対して製造販売承認がされた時点とは、状況が変化しているということです。医薬品は、あくまでその時代時代の医療の中で生み出され、活用される物ですので、「1回承認したらそれで終わり」というものではなく、必要がある時に、改めて評価する必要があります。このための制度を「再評価制度」といいます。
以下、それぞれについて見ていきましょう。
⑵ 新医薬品等の再審査制度(薬機法第14条の4)
① 再審査制度の流れ
新医薬品などの再審査制度は、以下の流れで行われます。
ⅰ 新医薬品などの製造販売承認時
⇒厚生労働大臣:再審査の指定(医薬品の種類に応じた調査期間の設定)
…下記②
ⅱ 製造販売(市場流通)の開始後
⇒製造販売業者:GPSPに従った申請データの収集
…下記③
⇒製造販売業者:厚生労働大臣に対する安全性定期報告
…下記④
ⅲ 調査期間経過から一定期間
⇒製造販売業者:厚生労働大臣(実際にはPMDA)に対する再審査の申請
…下記⑤
⇒PMDA :再審査の実施
…下記⑤
⇒厚生労働大臣:薬事・食品衛生審議会の諮問を経て、結果の決定
…下記⑥
⇒厚生労働大臣:再審査の結果の公表
…下記⑥
つまり、新薬などに関しては、最初から事後的な再確認(審査)が必要なことが分かっていますので、製造販売承認時において、再審査の指示と、医薬品の種類に応じた調査期間が設定されるものとされています。この場合、製造販売業者は、その調査期間中は、医薬品に関する情報収集を行い、定期的にその内容を報告します。そして、調査期間終了後に、収集されたデータを添付して再審査の申請を行い、改めて審査が行われるという流れです。
以下、各フローについてポイントをご説明します。
② 再審査の指定
新薬などに関しては、薬事承認の際に、厚生労働大臣から製造販売業者に対して再審査が指定されます。この際に、医薬品の種類に応じて調査期間が併せて指定されます(薬機法第14条の4第1項。ただし、緊急承認の際は別途条件と期限が設定されますので除外されます。)。
調査期間は、令和2年8月31日付の厚生労働省の通達(薬生薬審発0831第16号)『再審査機関の取扱いについて』により、詳細に定められています。その内容は、以下のとおりですが、この期間は、症例が少ないなどの情報収集の難しさなども考慮して設定されておりますが、「7」の「6年」を基準に長短が決められています。
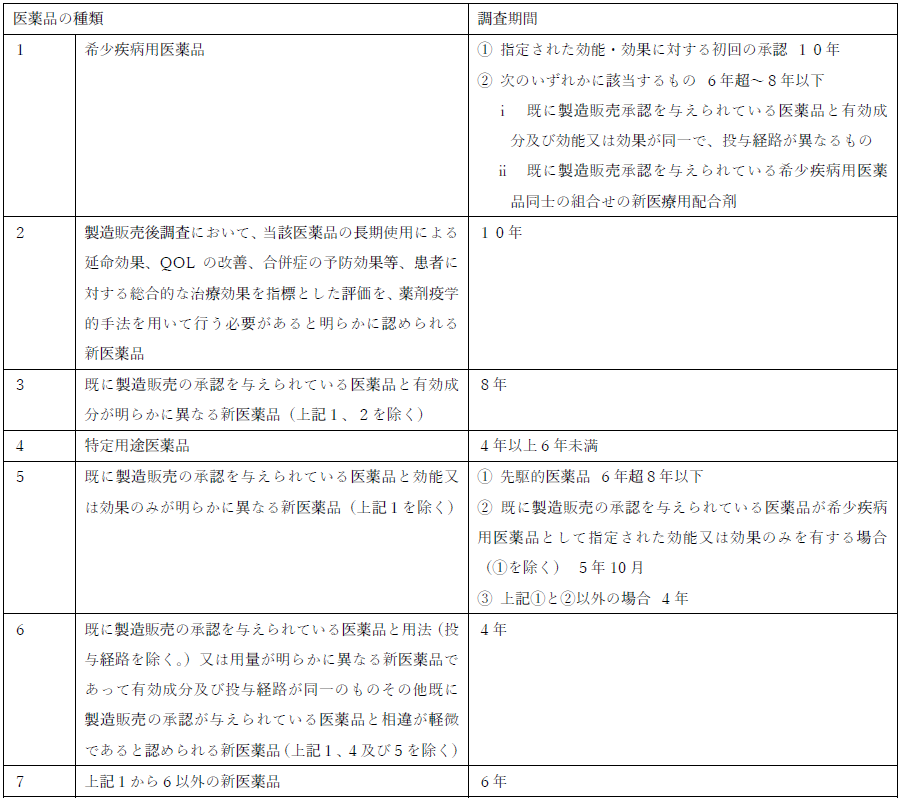
③ 再審査資料(申請データ)
上記の調査期間において、製造販売業者は新薬に関する情報を収集していくことになります。そして、最終的に再審査資料として整理されますが、要求される資料はGPSP省令(後述)に基づいて収集・作成されます(薬機法第14条の4第5項)。その内容は多岐に渡りますが、安全性に関する情報と有効性に関する情報がその中心です。
以下の項目は、安全性と有効性に関して、それぞれ資料とする事項になります。製造販売業者は単に情報を収集するだけでなく、問題点に対する措置や研究の実施なども求められることが分かります。
◎安全性に関する事項
1. 副作用・感染症発現状況
2. 追加の医薬品安全性監視計画の実施結果
3. 安全性に関する措置
4. 安全性に関する研究報告
5. 特定の背景を有する患者への投与に関する情報
6. 追加のリスク最小化計画の実施結果
7. その他の安全性に関する事項
8. 安全性検討事項及びその他の安全性に関する考察
◎有効性に関する事項
1. 有効性に関する調査・試験の実施結果
2. 有効性に関する措置
3. 有効性に関する研究報告
4. その他の有効性に関する事項
5. 有効性に関する検討事項及びその他の有効性に関する考察
④ 安全性定期報告
薬機法第14条の4第7項は「当該医薬品の使用の成績に関する調査その他厚生労働省令で定める調査を行い、その結果を厚生労働大臣に報告しなければならない。」としています。この「調査」は、略して「使用成績調査等」といわれます。
ここで得られる情報は、特に副作用については極めて重要なものになるため、定期的に厚生労働大臣に報告すべきものとされています。これを「安全性定期報告」といいます。
これについては薬機法施行規則第63条に詳細が定められており、原則として調査期間の開始日から2年間は半年以内ごとに、それ以降は1年ごとに行うものとされています。
⑤ 再審査の申請、再審査の実施
調査期間経過から3か月以内に、製造販売業者は、医薬品再審査申請書再審査資料をPMDAに提出し、再審査の申請を行います。
PMDAでは、GPSP省令に基づき、適合性調査、全般的事項、有効性、安全性に関する調査、審議などを行います。その後、厚生労働大臣に審議結果が伝えられ、食品衛生審議会の諮問を経ます。
⑥ 厚生労働大臣による結果の決定及び公表
上記の審査は、医薬品の有効性、つまり承認を受けた効能効果があるかどうか、安全性の事項と相対的に評価した際に、有効性を上回る有害性がないかどうかなどに関して行われます。
そのため、審査の結果、特に問題がない場合は、何の措置もなされません。
これに対し、薬機法第14条第2項に定める事由、つまり、薬事承認の際の不承認事由に該当する場合は、当然の結果として製造販売承認が取り消されます。この場合、販売の中止や製品の回収がなされます。
一方、問題はあるものの、承認事項の一部変更で対応できる場合は、製造販売承認事項の一部変更が命じられます。この場合は、効能効果に関する表示等の変更が必要になりますので、製造販売業者は速やかに対応することになります。
また、この結果は公表されます。
⑶ 医薬品の再評価制度(薬機法第14条の6)
① 再評価制度の歴史と現在の制度
再評価制度は、今から50年ほど前に始まった制度で、その後、当時の薬事法への明文化なども挟みながら、過去に承認されたあらゆる医療用医薬品、一般用医薬品を対象に行われました(第一次再評価、第二次再評価)。この当時は、一度薬事承認を受けた医薬品を見直す機会がそれまでになかったことから、その時点での再評価を行うことに主眼が置かれていました。もっとも、安全性・有効性に関する医薬品の問題点はその後も起こりうることから、より一般的な制度としてつくられたのが現在の再評価制度のうちの「薬効再評価」制度になります。
また、1997年からは主にジェネリック医薬品を対象に、品質の再評価制度が設けられました。これを「品質再評価」といいます。ジェネリック医薬品は、先発医薬品と同じ効能効果があるとされる意味では、安全性、有効性については同様であることから、専ら品質(製造による部分が大きいため)に関して再評価を行うという制度になっています。
つまり、現在の再評価制度は、薬品の3つの要素(有効性、安全性、品質)のうちの、有効性・安全性などを再評価する「薬効再評価」と、品質(溶出性)を再評価する「品質再評価」があることになります。
なお、品質再評価は、品質について再評価するものであり、安全性や有効性のように改めて人に対する治験を行う必要がありません。そして、先発医薬品を飲んだ時に腸から吸収される成分量と、ジェネリック医薬品を飲んだときに吸収される成分量が同じであれば、品質には問題がないと考えられます。このように、吸収される成分量を基準に双方が同じであることを「生物学的に同等」といい、具体的には、血液中の有効成分の濃度が同等であることになります。このためには、人体ではなく、溶出試験器、液体クロマトグラフ、紫外可視分光光度計などを使用して行う「溶出試験法」を実施します。
② 再評価制度の流れ
この医薬品の再評価制度は、以下の流れで行われます。
ⅰ 緊急の問題が発生した時など
⇒厚生労働大臣:再評価の範囲の指定と公示
ⅱ 再評価の範囲の公示後
⇒製造販売業者: GPSPに従った申請データの収集
⇒製造販売業者:厚生労働大臣(実際にはPMDA)に対する再評価の申請
⇒PMDA :再評価の審査の実施
⇒厚生労働大臣:薬事・食品衛生審議会の諮問を経て、結果の決定
⇒厚生労働大臣:再審査の結果の公表
再評価制度は既に薬事承認を受けた医薬品について、必要があれば再評価を行う制度のため、再審査制度とは異なり、厚生労働大臣による範囲の指定から始まります。しかも、この範囲の指定は特定の医薬品を対象とするのではなく、ある成分を含有する医薬品の全てを対象とすることも異なります。このため、範囲に含まれる医薬品を製造販売する業者の全てが対応する必要があります。
もっとも、その点を除けば、基本的な手続の流れは、再審査制度と同様です。
⑷ GPSP省令
① GPSP省令とは
医薬品の安全性、有効性、品質については、薬事承認申請においても必要になるものであり、前回は、その際には治験などの様々な試験結果を資料として添付する必要があることをご説明しました。
この点、再審査制度、再評価制度ともに、医薬品の安全性、有効性、品質について審査をするという意味では、承認申請時と共通しますが、ここまでご説明したとおり、市場流通後の制度では、市場で実際に使用されている状況に関する調査や、承認時には必要がないような試験が必要になることもあり、この点は薬事承認申請の時点とは大きく異なります。そのため、薬事承認の際のGLP省令やGCP省令とは対象が違います。
また、今回ご説明したGVP省令は、安全管理のための体制整備を目的とするものであり、その体制で行われる調査や試験に関する基準ではありません。
そのため、厚生労働省は、市場流通後の調査や試験に関する基準として、
「医薬品の製造販売後の調査及び試験の実施の基準に関する省令」
=(通称「GPSP省令」)
を定めています。
② GPSP省令の内容
「GPSP」というのは、「Good Post-marketing Study Practice」の略です。そのため、「PS」というのは「製造販売後(市場流通後)のStudy(研究)」という意味の「Post-marketing Study」という意味なので、こちらも他の省令との区別は容易かと思います。
なお、ややこしいのは、一般的に製造販売後調査のことを「PMS」と呼び、こちらは「Postmarketing Surveillance」の略なのですが、分かりやすくいえば、PSのなかにPMSがあるという関係になります。
そして、GPSP省令が定める「研究」とは、以下のような構造になっています。
製造販売後調査等(製造販売後のStudy)
① 使用成績評価
ⅰ 一般使用成績評価
ⅱ 特定使用成績評価
ⅲ 使用成績比較調査
② 製造販売後データベース調査
③ 製造販売後臨床試験
以下、各Studyについて見ていきましょう。
ⅰ 使用成績評価
使用成績評価は、GPSP省令第2条第1項第1号によると、
医療機関から収集した情報を用いて、診療において、医薬品の副作用による疾病等の種類別の発現状況並びに品質、有効性及び安全性に関する情報の検出又は確認のために行う調査であって、次に掲げるものをいう。以下同じ。)
イ 一般使用成績調査(医薬品を使用する者の条件を定めることなく行う調査(ハに規定する使用成績比較調査に該当するものを除く。)をいう。)
ロ 特定使用成績調査(小児、高齢者、妊産婦、腎機能障害又は肝機能障害を有する者、医薬品を長期に使用する者その他医薬品を使用する者の条件を定めて行う調査(ハに規定する使用成績比較調査に該当するものを除く。)をいう。)
ハ 使用成績比較調査(特定の医薬品を使用する者の情報と当該医薬品を使用しない者の情報とを比較することによって行う調査をいう。)
とされていますので、分かりにくいのですが、簡単に言えば、「条件を定めることなく」、つまり、臨床試験ではなく一般的な診療の中で処方されるなどして使用される場面での調査ということになります。
ⅱ 製造販売後データベース調査
製造販売後データベース調査は、GPSP省令第2条第1項第2号に
医療情報データベース取扱事業者が提供する医療情報データベースを用い、医薬品の副作用による疾病等の種類別の発現状況並びに品質、有効性及び安全性に関する情報の検出又は確認のために行う調査
とあるように、データベースから情報を収集する方法です。
ⅲ 製造販売後臨床試験
最後に、製造販売後臨床試験は、GPSP省令第2条第1項第3号に
治験、使用成績調査若しくは製造販売後データベース調査の成績に関する検討を行った結果得られた推定等を検証し、又は診療においては得られない品質、有効性及び安全性に関する情報を収集するため、医薬品について法第14条第1項若しくは第13項(法第19条の2第5項において準用する場合を含む。)又は第19条の2第1項の承認に係る用法、用量、効能及び効果に従い行う試験
とあります。分かりやすく言えば、一定の計画のもとに行われる試験のことです。この試験には、動物を対象とするもの、人を対象とするものなどがあるのは薬事承認申請の時と同様です。
6.まとめ
以上、今回は、医薬品の製造販売後の制度について、広く見て参りました。
かなりの文量になりましたが、医薬品特有の問題点を念頭に、安全性、有効性、品質に問題が生じないように薬機法が制度を設けているという大枠を踏まえて各制度を位置付ければ、理解が難しいこともなく、合理的な制度になっていることがお分かりいただけるかと思います。
・医薬品には、製造販売後も安全性、有効性、品質に関して問題がある
・安全管理・有効性の再確認のための前提として「製造販売後調査」がある
特に、安全管理のために、製造販売業者、医療機関、患者などから幅広く情報を収集する
・安全性、有効性ともに収集された情報をもとに、厚生労働省が必要な措置を行い、医薬品を管理する
・安全管理のための製造販売業者の体制に関する基準が「GVP省令」
有効性の再確認のための資料に関する基準が「GPSP省令」
これまで5回に亘って医薬品に関するルールをご説明して参りましたが、次回は、医薬品の最後の回として、消費者に対して医薬品の情報が表示される場面として、医薬品の容器などへの表示、添付文書、そして医薬品の広告に関する概要を解説します。








