

執筆:弁護士 早崎 智久(メディカル・ビューティー・ヘルスケアチーム)
連載:薬機法とは ~薬機法の基本~
『第1回 薬機法の全体像』はこちらから
『第2回 「医薬品等」とは』はこちらから
『第3回 医薬品の販売と薬局 ① -医薬品の種類-』はこちらから
『第4回 医薬品の販売と薬局② -医薬品の販売と薬局-』はこちらから
『第5回 医薬品の製造販売① -全体像と製造販売業・製造業・製造管理-』はこちらから
『第6回 医薬品の製造販売② -医薬品の製造販売承認-』はこちらから
『第7回 製造販売後の医薬品 -医薬品の安全管理と有効性の再確認-』はこちらから
『第8回 医薬品情報の消費者への表示-医薬品の表示、添付文書、広告-』はこちらから
- 1.はじめに
- 2.医薬品の製造販売承認の概要
- ⑴ 医薬品を市場に流通させるための概要(復習)
- ⑵ 医薬品の製造販売承認の意味
- 3.医薬品の製造販売承認を得るために必要な事項
- 4.医薬品の製造販売承認申請に必要なデータ
- ⑴ 承認申請に必要なデータ(総論)
- ⑵ 承認申請に必要なデータ(各論)
- 5.データの信頼性
- ⑴ 概要
- ⑵ 適切な試験①:非臨床試験
- ⑶ 適切な試験②:臨床試験(治験)
- 6.医薬品の製造販売承認の申請フロー
- ⑴ 承認申請
- ⑵ 承認審査
- ⑶ 審査報告書の作成・提出
- ⑷ 薬事・食品衛生審議会の関与
- ⑸ 厚生労働大臣の承認・却下の決定
- 7.承認期間を短縮化させるための諸制度
- ⑴ 標準的事務処理期間
- ⑵ 特例承認
- ⑶ 先駆け審査指定制度
- ⑷ 医薬品条件付き早期承認制度
- ⑸ 緊急承認
- 8.承認不要医薬品
- 9.外国の医薬品の製造販売
- 10.その他の重要事項
- ⑴ 承認事項一部変更承認(一変承認)
- ⑵ 薬事承認の承継
- 11.まとめ

1.はじめに
GVA法律事務所では、メディカル、美容、ヘルスケア領域に関して専門チームを設け、各分野について多様なサポートをさせていただいております。
薬機法の基本に関する連載5回目は、医薬品を市場に流通させることに関する規制のうち、事業者に対する規制内容に関してご説明しました。6回目となる今回は、医薬品自体に対する製造販売承認について解説いたします。
2.医薬品の製造販売承認の概要
⑴ 医薬品を市場に流通させるための概要(復習)
前回も、お話しましたように、「医薬品は、作りたい、輸入したいと思っても、自由に製造したり、輸入したりできない」ということがポイントです。
医薬品は食品と比較しても人体への影響が大きい(副作用など)ため、厳しい規制がかけられています。様々な試験を経た結果、「これなら医薬品として流通させてもよろしい」とならなければ、作ったり、輸入すること自体が許されないものになります。
前回、医薬品を市場に流通させるためには、その事業者において、製造販売業の許可を取らなければならず、また、製造するためには製造業許可が必要になることをお話しましたが、医薬品自体に対する製造販売承認はこれらとは別のものです。
医薬品については、
① 医薬品自体の製造販売に対する承認
② 医薬品を製造販売しようとする事業者に対する許可
の2つの規制があることを再度ご確認ください。
そして、この②は前回ご説明しましたので、今回は、①についてご説明いたします。
なお、「製造販売」という用語が、製造でも販売でもないことは、前回、「製造販売」業に関してご説明しましたが、この点は、医薬品の「製造販売」承認についても同じです。要するに、ある医薬品を市場に流通させるための承認、ということになります。
⑵ 医薬品の製造販売承認の意味
こちらも前回ご説明したことですが、医薬品は通常のモノと比べ、人体への影響が大きく、薬害により、多くの方々の生命や身体を傷つけてしまうような不幸な事件も発生しました。また、何の効き目もない医薬品(効能効果の無い医薬品)や品質の悪い医薬品も問題です。
そのため、医薬品には、安全性、効能効果、品質の全てにおいて、問題のないことが求められ、そのような製品でなければ、市場に流通させることはできません。
医薬品自体について、製造販売承認をする制度があるのは、このような問題が生じないことをデータに基づいて確認し、クリアした製品のみを市場に流通させるための制度になります。
※なお、ごく例外的な医薬品については、製造販売承認を必要としないものがあります。
3.医薬品の製造販売承認を得るために必要な事項
医薬品の製造販売承認は、薬機法の第14条に定められており、第2項において、承認が与えられない場合が列挙されています。
一 申請者が、第十二条第一項の許可(申請をした品目の種類に応じた許可に限る。)を受けていないとき。
二 申請に係る医薬品、医薬部外品又は化粧品を製造する製造所が、第十三条第一項の許可(申請をした品目について製造ができる区分に係るものに限る。)、第十三条の三第一項の認定(申請をした品目について製造ができる区分に係るものに限る。)又は第十三条の二の二第一項若しくは前条第一項の登録を受けていないとき。
三 申請に係る医薬品、医薬部外品又は化粧品の名称、成分、分量、用法、用量、効能、効果、副作用その他の品質、有効性及び安全性に関する事項の審査の結果、その物が次のイからハまでのいずれかに該当するとき。
イ 申請に係る医薬品又は医薬部外品が、その申請に係る効能又は効果を有すると認められないとき。
ロ 申請に係る医薬品又は医薬部外品が、その効能又は効果に比して著しく有害な作用を有することにより、医薬品又は医薬部外品として使用価値がないと認められるとき。
ハ イ又はロに掲げる場合のほか、医薬品、医薬部外品又は化粧品として不適当なものとして厚生労働省令で定める場合に該当するとき。
四 申請に係る医薬品、医薬部外品又は化粧品が政令で定めるものであるときは、その物の製造所における製造管理又は品質管理の方法が、厚生労働省令で定める基準に適合していると認められないとき。薬機法では、このように「承認を与えられない場合」という書き方をしていて、また、条文の引用がありますが、これを分かりやすくすると、以下のとおりになります。
① 承認申請をする事業者が製造販売業許可を取得していること
② 医薬品を製造する製造所が製造業許可などを取得していること
③ 医薬品の効能効果、安全性、品質において問題がないこと
④ 品質管理、製造管理についてGQP、GMPの基準を満たすこと
こうしてみると、①、②、④は、前回ご説明した事項に関わり、主に事業者に関する事項になりますので、医薬品の製造販売承認における医薬品自体に関するものとして特にポイントになるのは、③の「医薬品の効能効果、安全性、品質において問題がないこと」になります。
そこで、次は、この③を証明するために必要な資料(データ)について見ていきます。
4.医薬品の製造販売承認申請に必要なデータ
⑴ 承認申請に必要なデータ(総論)
承認申請に必要な資料(データ)については、薬機法施行規則第40条1項1号と厚生労働省の通達(平成26年11月21日薬食発1121第2号「医薬品の承認申請について」で以下のように定められています。
「イ」~「チ」まで全8つの項目に分けられ、それぞれの右の欄にあるように、さらに細かく分類された資料(データ)の提出が必要になります。
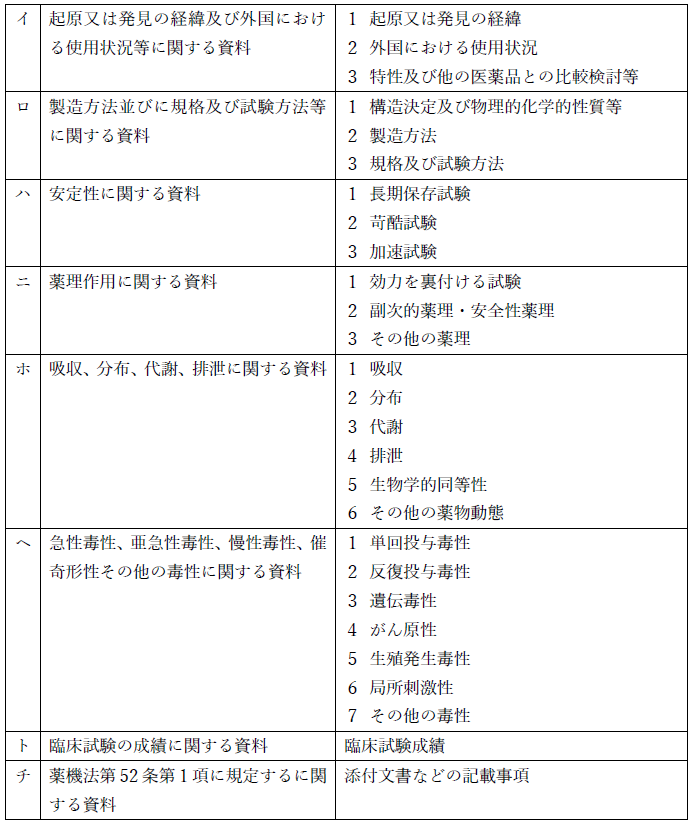
こちらは、求められている資料の項目を是非とも一つ一つ見ていただきたいです。承認を求める医薬品の概要、作り方、品質、効能効果、安全性(副作用)、そして試験結果から添付文書の記載内容まで、網羅的に審査対象となっていることが分かります。
医薬品は、ここまで内容を精査されたうえで市場に流通していることが分かりますし、裏返せば、ここまで精査を受けないと、市場に流通させることはできないとも言えます。
⑵ 承認申請に必要なデータ(各論)
上の⑴では、医薬品の承認申請に必要な資料の全体を見ましたが、医薬品には様々な種類がありますので、全ての医薬品について、これらの資料が全て必要になるわけではありません。
本連載の第3回目で医薬品の種類をご説明し、大きく「医療用医薬品」と「一般用医薬品(市販薬、OTC医薬品)」に分かれることを見て参りましたが、医薬品の承認申請でも、大きくはこの二つに分かれています。
① 医療用医薬品の承認申請区分ごとに必要なデータ
まず、医療用医薬品についてですが、以下の表のとおりとなっています。この区分は、厚生労働省の通達である「医薬品の承認申請について」(平成26年11月21日薬食発1121第2号通知)において定められています。
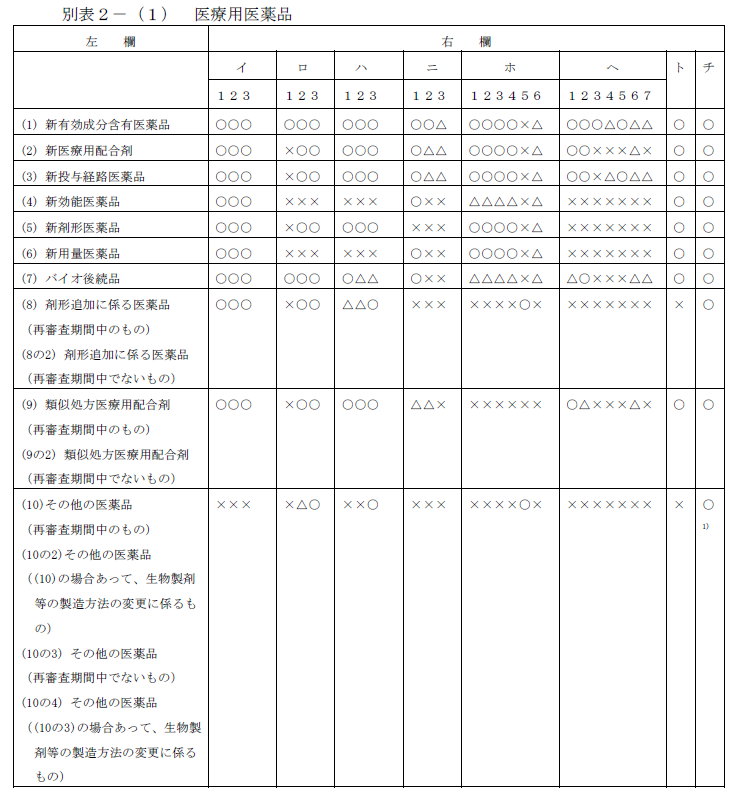
この区分を見ると、医療用医薬品については10種類のカテゴリに分けられていることが分かります。この区分は、承認申請という観点から分けられているものです。
上記「⑴」でどのようなデータが必要になるのかの概要を見ましたが、医薬品には、世界で初めて作られるような全く新しいものがある一方で、別会社が既に製造販売している医薬品と同じ成分で同様の効能効果を有するもの、同じ成分で同様の効能効果の医薬品でも形状が異なるもの(粒、カプセルなど)など、様々なものがあります。そして、例えば全く同じ成分であれば、改めて全部を審査する必要もありません。そのため、その種類により、必要となるデータも異なってきます。
なお、この表にある「イ」から「チ」は、上記「⑴」の表に対応しています。また、「〇」とあるのはデータが必要なもの、「✕」とあるのは不要なもの、「△」は医薬品によっては必要になるものという意味です。
そのため、例えば「新効能医薬品」については、「ロ 製造方法並びに規格及び試験方法等に関する資料」「ハ 安定性に関する資料に関するデータ」「ヘ 急性毒性、亜急性毒性、慢性毒性、催奇形性その他の毒性に関する資料」は、いずれも「✕」とあるため、データの提出は不要となります。
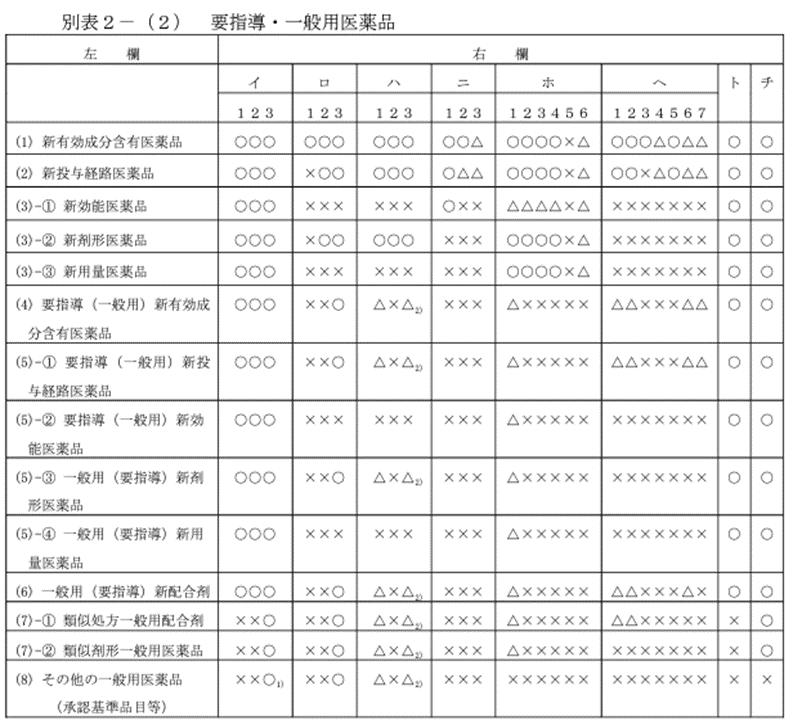
② 要指導・一般用医薬品の承認申請区分ごとに必要なデータ
要指導・一般用医薬品も、考え方は同じです。医薬品の種類に応じて、必要なデータが変わります。
5.データの信頼性
⑴ 概要
上記で見てきたように、医薬品の承認申請には、その医薬品ごとに必要な資料が分けられていることが分かったと思います。ただし、承認申請は、医薬品の有効性、安全性、品質をチェックするために行われるものなので、適当な資料が提出されても、チェックはできません。このデータ(申請資料)の信頼性については、薬機法施行規則の第43条が次のように定めています。
(申請資料の信頼性の基準)
第43条 法第14条第3項後段(同条第15項において準用する場合及び法第14条の2の25項の規定により読み替えて適用される場合を含む。)に規定する資料は、医薬品の安全性に関する非臨床試験の実施の基準に関する省令(平成9年厚生省令第21号)、医薬品の臨床試験の実施の基準に関する省令(平成9年厚生省令第28号)及び医薬品の製造販売後の調査及び試験の実施の基準に関する省令(平成16年厚生労働省令第171号)に定めるもののほか、次に掲げるところにより、収集され、かつ、作成されたものでなければならない。
1 当該資料は、これを作成することを目的として行われた調査又は試験において得られた結果に基づき正確に作成されたものであること。
2 前号の調査又は試験において、申請に係る医薬品についてその申請に係る品質、有効性又は安全性を有することを疑わせる調査結果、試験成績等が得られた場合には、当該調査結果、試験成績等についても検討及び評価が行われ、その結果が当該資料に記載されていること。
3 当該資料の根拠になった資料は、法第14条第1項又は第15項の承認(法第14条の2の2第1項の規定により条件及び期限を付したものを除く。)を与える又は与えない旨の処分の日まで保存されていること。ただし、資料の性質上その保存が著しく困難であると認められるものにあっては、この限りでない。
これを整理すると、以下のようになります。
Ⅰ 申請資料が、3つの省令に従って倫理的、科学的に適切に実施された試験の成績に基づいていること
※3つの省令
ⅰ 医薬品の安全性に関する非臨床試験の実施の基準に関する省令
ⅱ 医薬品の臨床試験の実施の基準に関する省令
ⅲ 医薬品の製造販売後の調査及び試験の実施の基準に関する省令
Ⅱ 申請資料が、「申請資料の信頼性の基準」に従って、試験結果に基づいて適切かつ正確に作成されていること
※「申請資料の信頼性の基準」
ⅰ 正確性
生のデータに基づき、正確に申請資料が作成されていること
ⅱ 完全性・網羅性
都合の悪いデータを含むすべてのデータが申請資料に記載されていること
ⅲ データの保存
承認の可否が判断されるまで、生のデータが保存されていること
つまり、まとめると、「適切に行われた試験の結果が、正確かつ完全に記載された資料であり、きちんと保存されていることが必要」ということになります。
なお、このうちのⅡについては、上に記載した内容のとおりですので、次は、特に重要な「Ⅰ」の「適切な試験」という点について、より詳細に見ていきましょう。
⑵ 適切な試験①:非臨床試験
まず、「非臨床試験」というのは、動物試験などの人を対象にしない試験のことです。そして、この試験の詳細を定めているのが、
「医薬品の安全性に関する非臨床試験の実施の基準に関する省令」
=通称:GLP省令(Good Laboratory Practice)
になります。
この「L」はLaboratory(=研究室)の頭文字ですが、この次にご説明する臨床試験が人を対象にしたものであり、主に病院等の医療機関(Clinic)で行われることと対比すると、非臨床試験の基準が、研究室の「L」というのは分かりやすいかと思います。
GLPでは、非臨床試験の管理の方法が詳細に定められており、概要は以下のようになります。
・試験を行う職員や組織に関する事項
・試験を行う施設や機器に関する事項
・機器の操作手順書(SOP)
・動物の飼育管理
・被験物質などに関する事項
・試験の計画、実施に関する事項
・結果の報告や保存に関する事項
人を対象にする臨床試験では、試験自体に一定の危険を伴うものですので、その安全性を判断するためにも、非臨床試験が不可欠です。そのため、非臨床試験は、次の臨床試験を行う前に実施されるものになります。
⑶ 適切な試験②:臨床試験(治験)
非臨床試験を経て、一定の安全性が確認できたら、人を対象にした試験が行われます。これを「臨床試験」といい、特に、医薬品等の製造販売承認申請に必要なデータを収集する目的で行われる臨床試験のことを「治験(ちけん)」(薬機法第2条17号)といいます。
この治験に関しては、薬機法では、医薬品の製造販売承認に関する一連の条項からはかなり離れた第17章「雑則」のなかに、第80条の2と第80条の3として定められています。
この中で重要な事項としては、
⑴ 厚生労働省令として、治験に関する基準が定められていること
⑵ 治験には、①製薬メーカーが医師に対して治験の依頼をする場合と②医師が自ら治験を実施する場合の2パターンがあること
⑶ 治験の実施には、厚生労働大臣への事前の治験計画の届出が必要なこと
⑷ 届出から30日が経過しないと治験は実施できないこと
があります。
この内、⑴の治験に関する基準が、
「医薬品の臨床試験の実施の基準に関する省令」
=通称:GCP省令(Good Clinical Practice)
になります。この「C」はClinical(=臨床の、診療所の)の頭文字であり、治験は、病院等の医療機関により、医師によって行われます。
GCPの目次を見ると、以下のようになります。
第一章 総則(第一条―第三条)
第二章 治験の準備に関する基準
第一節 治験の依頼をしようとする者による治験の準備に関する基準(第四条―第十五条)
第二節 自ら治験を実施しようとする者による治験の準備に関する基準(第十五条の二―第十五条の九)
第三章 治験の管理に関する基準
第一節 治験依頼者による治験の管理に関する基準(第十六条―第二十六条)
第二節 自ら治験を実施する者による治験の管理に関する基準(第二十六条の二―第二十六条の十二)
第四章 治験を行う基準(第二十七条―第五十五条)
第一節 治験審査委員会(第二十七条―第三十四条)
第二節 実施医療機関(第三十五条―第四十一条)
第三節 治験責任医師(第四十二条―第四十九条)
第四節 被験者の同意(第五十条―第五十五条)
第五章 再審査等の資料の基準(第五十六条)
第六章 治験の依頼等の基準(第五十七条―第五十九条)
附則
詳細な説明は割愛しますが、治験については、準備、管理、実施、そして、製薬メーカーからの医師への依頼に関する事項まで、詳細にルールが定められています。
6.医薬品の製造販売承認の申請フロー
おさらいになりますが、医薬品の製造販売の承認を得るためには、
① 承認申請をする事業者が製造販売業許可を取得していること
② 医薬品を製造する製造所が製造業許可などを取得していること
③ 医薬品の効能効果、安全性、品質において問題がないこと
④ 品質管理、製造管理についてGQP、GMPの基準を満たすことという4つの要件をクリアする必要があります。そして、③を証明するために、多数の資料(データ)が必要になり、データは、GLPとGCPが定める基準に従った試験結果に基づく必要があります。
そのため、ある医薬品の製造販売をするために、メーカー(製造販売業者)は、時間をかけて試験を行い、データを集めていきます。
そのようにしてデータが揃ったら、いよいよ医薬品として市場に流通させるための承認をもらうために、申請が行われます。これが医薬品の製造販売承認申請であり、「薬事申請」(=RA)とも言われます。
以下では、承認手続のフローに沿って、内容を見ていきます。
⑴ 承認申請
医薬品の製造販売承認を行うのは、厚生労働大臣です。そのため、承認申請も厚生労働省に提出することで行います。なお、一般用医薬品の一部については、都道府県知事が処理するとされているものがあります(薬機法第81条、一般用医薬品の承認区分の⑻)。この場合の提出先は都道府県になります。
⑵ 承認審査
薬機法第14条の2は、「厚生労働大臣は、機構に、医薬品…の前条の承認のための審査並びに同条第5項及び第6項…の規定による調査…を行わせることができる。」としています。
ここで「機構」とされているのが、独立行政法人医薬品医療機器総合機構であり、略称の「PMDA(ピーエムディーエー)」で呼ばれることが多いです。また、条文では「行わせることができる」とありますが、基本的に、ほぼすべての承認審査はPMDAが行っています。
※なお、PMDAでは、申請に先立つ事前の相談も受け付けており、一般的にも、申請前の相談を経たほうが、効率的に審査を行うことができます。
この審査は、PMDAが組成した専門家チームで行われますが、後でご説明する薬事・食品衛生審議会の薬事分科会の専門委員が助言をします。
専門化チームは、申請者が提出した書類を審査するだけでなく面接における質問も行い、問題点を整理し、これに基づき指示がなされ、協議の上で行われていきます。
また、PMDAでは、薬機法第14条第6項による適合性の調査(資料が、厚生労働省令で定める基準に従って収集され、かつ、作成されたものかどうかの調査です。)と、同条第7項によるGMP省令への適合性の調査も並行して行います。
⑶ 審査報告書の作成・提出
この審査が終了すると、PMDAは、審査結果を「審査報告書」としてまとめ、厚生労働省に報告します。
⑷ 薬事・食品衛生審議会の関与
審査報告を受けた厚生労働省は、対象となる医薬品の種類に応じて、以下のような対応を行います。
① 薬事・食品衛生審議会の薬事分科会の意見を聞く必要があるものについては、同会に対して諮問をする。同会は、審議を行い、意見を厚生労働大臣に述べる。
② 報告で足りるとされているものについては、同会に対して報告をする。
③ 諮問・報告のいずれも不要とされているもの(後発医薬品など)については、事務局で審査する。
⑸ 厚生労働大臣の承認・却下の決定
厚生労働大臣は、上記⑷を経た後、これまでの審査や薬事分科会の意見を踏まえ、承認か却下をします。
承認となった場合は、晴れて医薬品として市場に流通させることが認められます。
7.承認期間を短縮化させるための諸制度
⑴ 標準的事務処理期間
以上のように、慎重な手続を経て審査は進められますが、医薬品の中には病に苦しむ人にとって救いとなるものもあり、あまりに審査に時間がかかってしまうと、問題も生じます。そのため、目安となる審査期間が定められています。正式には「標準的事務処理期間」と言いますが、一般的には通称の「タイムクロック(TC)」と呼ばれます。新医薬品の場合は1年とされています。
もっとも、医薬品の性質から、一定の期間がかかります。そのため、特殊な事情がある場合により迅速な承認ができるようにするための制度が設けられています。
⑵ 特例承認
特例承認は、緊急で他に適当な方法がない場合に、海外において、日本と同等水準の承認制度に基づいて先行して製造販売が認められた医薬品に関して、通常よりも簡略化された手続きで承認するための制度です。これは、日本では、日本での薬事承認を経ないと製造販売ができないものの、日本と同程度の慎重な審査を経ている以上は、これを信頼することで、タイムクロックを短縮させるものと言えます。
⑶ 先駆け審査指定制度
こちらは、平成27年の厚生労働省通知によって試行的に導入された制度ですが、令和元年の改正薬機法により正式に明文化されています(薬機法第77条の2第2項)。
この制度は、日本の大学等の基礎研究能力が高く、実用化できれば、世界で最先端の治療薬を生み出すことができると考えられているものの、諸外国と比較し、これを実用化する能力が低いとも指摘され、また、世界に先駆けて実用化しないと、特許等の知的財産権を取得できません。そこで、このような世界の先駆けになるような物について、優先的に審査をする優遇制度になります。
具体的な内容としては、厚生労働大臣が、先駆的医薬品、特定用途医薬品として指定することにより、医薬品メーカーは、優先相談、事前評価の充実のほか、タイムクロック目標を6か月とする優先審査等を内容とする優遇措置を受けることができます。
⑷ 医薬品条件付き早期承認制度
平成29年の厚生労働省通知によって試行的に導入された制度ですが、令和元年の改正薬機法により正式に明文化されています。
この制度は、臨床試験の実施が難しい場合のためのものです。つまり、通常、医薬品の用法・用量等の設定のためには、検証的臨床試験、つまり多数の患者に対象となる医薬品を投与・使用し、有効性・安全性を検討することが必要になります。しかし、患者数が少ない疾患に対する医薬品については、多数の患者に投与・使用することが困難であり、少なくとも長時間を要するものでした。もっとも、患者が少なくても、その方々に早く治療薬を届ける必要があります。
そのため、厚生労働大臣において、①医療上特にその必要性が高いと認められる場合で、②検証的臨床試験の実施が困難である等の場合という2つを満たす場合は、臨床試験の試験成績に関する資料の一部の提出を要しないとすることができます(ただし、検証的臨床試験以外の臨床試験等で一定程度の有効性・安全性を確認することが想定されています)。また、厚生労働大臣は、使用の成績に関する調査の実施、適正な使用の確保のために必要な措置の実施等の条件を付して、製造販売の承認をすることができるものとされます。そのため、この条件付きの承認を受けた製造販売業者等は、その使用の成績に関する資料等を厚生労働大臣に提出するものとされ、提出を受けた厚生労働大臣は、品質、有効性、安全性等の使用の成績に関する調査を行います。また、この調査結果を踏まえて、条件の変更や安全対策の実施等の適切な使用の確保のために必要な措置をとることができます。
⑸ 緊急承認
こちらは、本年令和4年の改正薬機法により創設された制度です。新型コロナウイルスへの対応についても、これまでは、上記⑴でご説明した「特例承認」制度を行ってきたものの、これよりも早く承認ができるようにすることで、より有効な感染症対策が行えるとの考えのもとで創設された制度です。
特例承認との違いを見ると分かりやすいのですが、特例承認が海外で流通している医薬品を対象にするのに対し、緊急承認はこの制限を外し、全ての医薬品を対象にします。また、医薬品の承認では、有効性と安全性の確認が不可欠ですが、本制度では、有効性については推定できればよいとしています。具体的には、臨床試験が完了していない場合でも、推定できれば、条件を付けた上での承認が可能となります。
8.承認不要医薬品
ごく例外的なものですが、日本薬局方に基準のある医薬品について、製造販売承認が不要とされています。一般的に「承認不要医薬品」と呼ばれるもので、厚生労働大臣が指定しており、平成6年厚生省告示第104号によって示されています。
9.外国の医薬品の製造販売
以上は、日本の医薬品を前提としたものですが、優れた医薬品は日本国内に限らず、世界中で製造販売されています。実際に、最近の新型コロナウイルスに関しても、ワクチンは外国製のものがほとんどです。このような外国製の医薬品は、どのように市場に流通するのでしょうか。
まず、最初でも触れた前提を思い出していただきたいのですが、日本の薬機法では、医薬品自体に対する製造販売承認と、事業者に対する製造販売業許可の2つが必要でした。この構造は、外国製の医薬品の場合も同様です。
この場合、原則通りに、外国の医薬品メーカーが日本法人を設立し、その日本法人が日本国内の企業として製造販売業許可を取得する方法もあります。また、日本の製造販売業者が、外国の医薬品メーカーからライセンスを受けて製造販売することもあります。
一方で、外国の医薬品メーカーが直接に承認を受けることのできる特別な方法も認められています。それが「選任外国製造医薬品等製造販売業者制度」と呼ばれるものです(薬機法第19条の2)。
この制度は、以下の手順で進められます。
① まず、日本国内で製造販売業者として当該医薬品について責任を担う者が必要になるので、外国の医薬品メーカーは、その医薬品の品目の種類に応じて既に製造販売業許可を得ている業者の中から、製造販売業者を選任します(選任製造販売業者)。
② 次に、外国の医薬品メーカーは、厚生労働省に対し、承認の申請をします。
③ 厚生労働省は、この申請を審査し、問題がないと判断すると、外国の医薬品メーカーに対して外国特例承認をすると共に、①の選任製造販売業者に対して、製造販売業許可を行います。
これにより、外国の医薬品が、選任製造販売業者により、日本国内に流通されることになります。
10.その他の重要事項
⑴ 承認事項一部変更承認(一変承認)
医薬品の製造販売には承認が必要なことを見てきましたが、承認後に、承認された事項の一部を変更する必要がある場合もあります。変更前の内容で承認を受けた以上は、その一部とはいえ変更する場合は、改めてその部分の承認を受ける必要があります(薬機法第14条第15項)。
⑵ 薬事承認の承継
医薬品の製造販売承認とは言っても、承認を受けるのは医薬品自体ではなく、その製造販売を求めるメーカーです。そして、そのメーカーも、新設合併、吸収合併などにより、別の法人になることがあり得ます。このような場合、再度の薬事承認を行うことは負担が大きく、また二度手間になるため、薬機法は、「薬事承認の承継」を認めています(薬機法第14条の8)。
11.まとめ
以上、今回は、医薬品自体に対する製造販売承認(薬事承認)について、見てきました。
分量も多いものですが、以下の点がポイントになります。
医薬品の薬事承認では、事業者に関する事項のほか、医薬品の効能効果、安全性、品質において問題がないことを証明する必要がある
承認申請に必要なデータは医薬品の区分により異なる
承認審査はPMDAで行われる
審査に必要な時間(タイムクロック)は1年程度だが、制度により、これより早期の承認を受けられる場合がある
外国の医薬品メーカーのための特別な制度がある
2回にわたり、医薬品を市場に流通させるための制度を見てきました。流通させた後の販売については既にご説明していますので、次回は、医薬品を市場に流通させた後の安全管理について解説します。
連載:薬機法とは ~薬機法の基本~
※過去のものはこちらです
『第1回 薬機法の全体像』はこちらから
『第2回 「医薬品等」とは』はこちらから
『第3回 医薬品の販売と薬局 ① -医薬品の種類-』はこちらから
『第4回 医薬品の販売と薬局② -医薬品の販売と薬局-』はこちらから
『第5回 医薬品の製造販売① -全体像と製造販売業・製造業・製造管理-』はこちらから
監修
弁護士 鈴木 景
(都内法律事務所からインハウスローヤーを経て、2017年GVA法律事務所入所。 スタートアップから大手上場企業まで、新規事業開発支援、契約書作成レビュー支援、株式による資金調達、M&AやIPOによるExitの支援など幅広く対応。 対応領域も、医療・美容に関する広告規制対応や、食品関連ビジネス、旅行関連ビジネス、NFT関連ビジネスと幅広い。)








